


※ iGPS INTRODUCTION:
The phosphorylation catalyzed by protein kinases (PKs) is one of the most important and ubiquitous post-translational modifications (PTMs) of proteins. It temporally and spatially modifies approximately 30% of all cellular proteins, and plays a crucial role in regulating a variety of biological processes, such as signal transduction and the cell cycle (Kobe, et al., 2005; Manning, et al., 2002; Olsen, et al., 2006; Ptacek, et al., 2005; Ptacek and Snyder, 2006; Ubersax and Ferrell, 2007). The human genome encodes 518 PK genes (~2% of the genome), with different PKs exhibiting distinct recognition specificities; each PK modifies only a limited subset of substrates, thus guaranteeing cell-signaling fidelity (Kobe, et al., 2005; Manning, et al., 2002; Olsen, et al., 2006; Ptacek, et al., 2005; Ptacek and Snyder, 2006; Ubersax and Ferrell, 2007). It is accepted that short linear motifs (SLMs) around phosphorylation sites (p-sites) provide the primary specificity (Hjerrild and Gammeltoft, 2006; Kobe, et al., 2005; Kreegipuu, et al., 1998; Songyang, et al., 1996>; Ubersax and Ferrell, 2007), while a variety of additional contextual factors, including co-localization, co-expression, co-complex, or physical interaction of the PKs with their targets, contribute additional specificity in vivo (Biondi and Nebreda, 2003; Holland and Cooper, 1999; Linding, et al., 2007; Linding, et al., 2008; Tan and Linding, 2009; Yaffe, et al., 2001). Aberrances of PKs or key substrates disrupt normal function, rewire signaling pathways, and are implicated in various diseases and cancers (Erxleben, et al., 2006; Gentile, et al., 2008; Manning, et al., 2002; Radivojac, et al., 2008; Ren, et al., 2010). In this regard, the identification of kinase-specific p-sites and the systematic elucidation of site-specific kinase-substrate relations (ssKSRs) would provide a fundamental basis for understanding cell plasticity and dynamics and for dissecting the molecular mechanisms of various diseases, while the ultimate progress could suggest potential drug targets for future biomedical design (Linding, et al., 2007; Linding, et al., 2008; Tan and Linding, 2009).
In this work, we developed a software package of iGPS (GPS algorithm with the interaction filter, or in vivo GPS) mainly for the prediction of in vivo ssKSRs. Eukaryotic PKs were classified into a hierarchy with four levels: group, family, subfamily, and single PK (Manning, et al., 2002). Based on the hypothesis that similar PKs recognize similar SLMs, we selected a predictor in GPS 2.0 (Xue, et al., 2008) for each PK and directly predicted the potential PKs for the non-annotated p-sites from the phosphoproteomic studies. Consequently, protein-protein interaction (PPI) information was used as the major contextual factor to filtrate potentially false-positive hits. The performance of iGPS was shown by critical evaluations and comparisons to be promising for the accurate prediction of in vivo ssKSRs. Based on the prediction results of iGPS, we modeled eukaryotic protein phosphorylation networks (PPNs) at different levels, including whole proteome, pathway and tissues/organs. By additionally computational analyses, we obtained a substantial number of potentially new observations, which can be subjected to further experimental manipulation. This study provides useful information for the understanding of the functional organization and diversity of eukaryotic phosphoproteomes at a systemic level and can be a model for analyzing other PTM-regulating proteomes.
The iGPS 1.0 is freely available at: http://igps.biocuckoo.org.
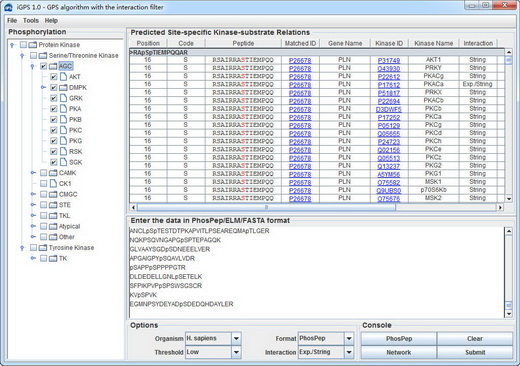
iGPS 1.0 User Interface
For publication of results please cite the following article: Systematic analysis of protein phosphorylation networks from phosphoproteomic data. Chunxia Song#, Mingliang Ye#, Xinning Jiang, Guanghui Han, Zhou Songyang, Yexiong Tan, Hongyang Wang, Jian Ren*, Yu Xue* & Hanfa Zou*. Mol Cell Proteomics. 2012 Jul 13. |